Collapse Kinetics and Chevron Plots from Simulations of Denaturant-Dependent Folding of Globular Proteins
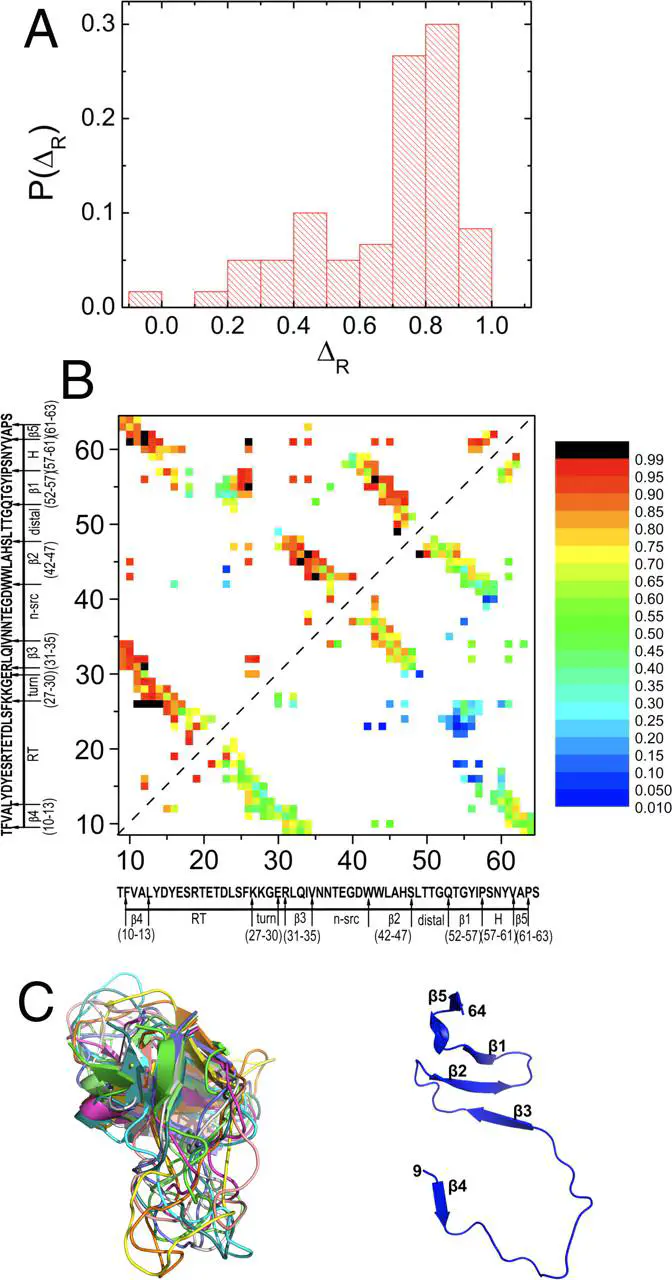 Image credit: G. Reddy
Image credit: G. Reddy
Abstract
Quantitative description of how proteins fold under experimental conditions remains a challenging problem. Experiments often use urea and guanidinium chloride to study folding whereas the natural variable in simulations is temperature. To bridge the gap, we use the molecular transfer model that combines measured denaturant-dependent transfer free energies for the peptide group and amino acid residues, and a coarse-grained Cα-side chain model for polypeptide chains to simulate the folding of src SH3 domain. Stability of the native state decreases linearly as [C] (the concentration of guanidinium chloride) increases with the slope, m, that is in excellent agreement with experiments. Remarkably, the calculated folding rate at [C] = 0 is only 16-fold larger than the measured value. Most importantly ln kobs (kobs is the sum of folding and unfolding rates) as a function of [C] has the characteristic V (chevron) shape. In every folding trajectory, the times for reaching the native state, interactions stabilizing all the substructures, and global collapse coincide. The value of (mf is the slope of the folding arm of the chevron plot) is identical to the fraction of buried solvent accessible surface area in the structures of the transition state ensemble. In the dominant transition state, which does not vary significantly at low [C], the core of the protein and certain loops are structured. Besides solving the long-standing problem of computing the chevron plot, our work lays the foundation for incorporating denaturant effects in a physically transparent manner either in all-atom or coarse-grained simulations.