Theory of the Molecular Transfer Model for Proteins with Applications to the Folding of the src-SH3 Domain
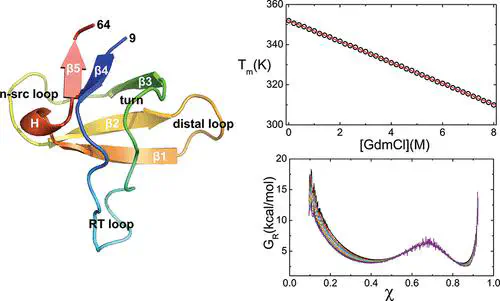 Image credit: G. Reddy
Image credit: G. Reddy
Abstract
A theoretical basis for the molecular transfer model (MTM), which takes into account the effects of denaturants by combining experimental data and molecular models for proteins, is provided. We show that the MTM is a mean field-like model that implicitly takes into account denaturant-induced many body interactions. The MTM in conjunction with the coarse-grained self organized polymer model with side chains (SOP-SC) for polypeptide chains is used to simulate the folding of the src-SH3 domain as a function of temperature (T) and guanidine hydrochloride (GdmCl) concentration [C]. Besides reproducing the thermodynamic aspects of SH3 folding, the SOP-SC also captures the cooperativity of the folding transitions. A number of experimentally testable predictions are also made. First, we predict that the melting temperature Tm([C]) decreases linearly as [C] increases. Second, we show that the midpoints Cm,i and melting temperatures Tm,i at which individual residues acquire 50% of their native contacts differ from the global midpoint (Cm ≈ 2.5 M) and melting temperature (Tm = 355 K) at which the folded and unfolded states coexist. Dispersion in Cm,i is greater than that found for Tm,i. Third, folding kinetics at [C] = 0 M shows that the acquisition of contacts between all the secondary structural elements and global folding occur nearly simultaneously. Finally, from the free energy profiles as a function of the structural overlap function and the radius of gyration of the protein, we find that at a fixed T the transition state moves toward the folded state as [C] increases in accord with the Hammond postulate. In contrast, we predict that along the locus of points Tm([C]) the location of the transition state does not change. The theory and the models used here are sufficiently general for studying the folding of other single domain proteins.