 Image credit: G. Reddy
Image credit: G. Reddy
Abstract
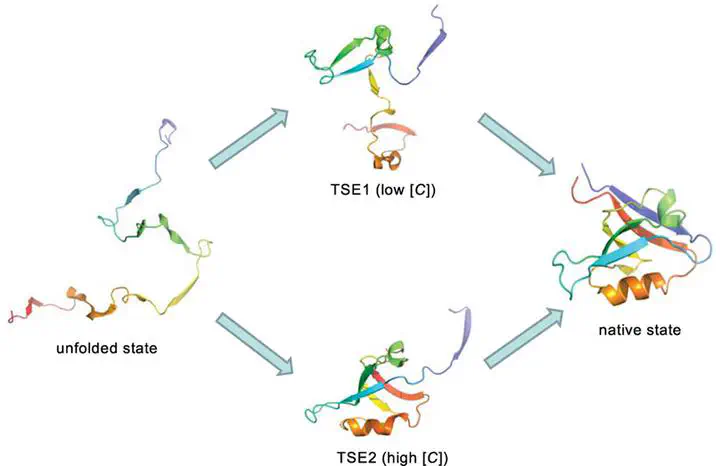
A major challenge in molecular simulations is to describe denaturant-dependent folding of proteins in order to make direct comparisons with in vitro experiments. We use the molecular transfer model (MTM), currently the only method that accomplishes this goal, albeit phenomenologically, to quantitatively describe urea-dependent folding of the PDZ2 domain, a protein that is important in molecular recognition and signaling. Experiments show that urea-dependent unfolding rates of the PDZ2 domain exhibit a downward curvature at high urea concentrations ([C]’s), which has been interpreted as indicating the presence of a sparsely populated high energy intermediate. Simulations using the MTM and a coarse-grained self-organized polymer (SOP) representation of PDZ2 are used to show that the intermediate (IEQ), which has some native-like character, is present in equilibrium both in the presence and absence of urea. The free energy profiles as a function of the structural overlap order parameter show that there are two barriers separating the folded and unfolded states. Structures of the transition state ensembles (TSE1 separating the unfolded and IEQ and TSE2 separating IEQ and the native state), determined using the Pfold method, show that TSE1 is greatly expanded while TSE2 is compact and native-like. Folding trajectories reveal that PDZ2 folds by parallel routes. In one pathway, folding occurs exclusively through I1, which resembles IEQ. In a fraction of trajectories, constituting the second pathway, folding occurs through a combination of I1 and a kinetic intermediate. We establish that the radius of gyration (RgU) of the unfolded state is more compact (by ∼9%) under native conditions. Theory and simulations show that the decrease in RgU occurs on the time scale on the order of at most ∼20 μs. The modest decrease in RgU and the rapid collapse suggest that high spatial and temporal resolution, currently beyond the scope of most small-angle X-ray scattering experiments, are needed to detect compaction in finite-sized proteins. The present work further establishes that MTM is efficacious in producing nearly quantitative predictions for folding of proteins under conditions used in experiments.