Role of Disulfide Bonds and Topological Frustration in the Kinetic Partitioning of Lysozyme Folding Pathways
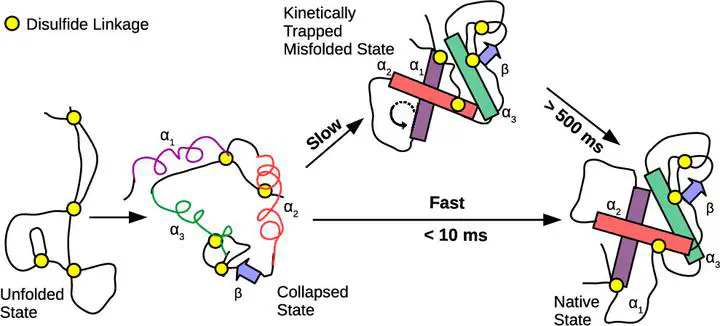 Image credit: G. Reddy
Image credit: G. Reddy
Abstract
Disulfide bonds in proteins can strongly influence the folding pathways by constraining the conformational space. Lysozyme has four disulfide bonds and is widely studied for its antibacterial properties. Experiments on lysozyme infer that the protein folds through a fast and a slow pathway. However, the reasons for the kinetic partitioning in the folding pathways are not completely clear. Using a coarse-grained protein model and simulations, we show that two out of the four disulfide bonds, which are present in the α-domain of lysozyme, are responsible for the slow folding pathway. In this pathway, a kinetically trapped intermediate state, which is close to the native state, is populated. In this state, the orientations of α-helices present in the α-domain are misaligned relative to each other. The protein in this state has to partially unfold by breaking down the interhelical contacts between the misaligned helices to fold to the native state. However, the topological constraints due to the two disulfide bonds present in the α-domain make the protein less flexible, and it is trapped in this conformation for hundreds of milliseconds. On disabling these disulfide bonds, we find that the kinetically trapped intermediate state and the slow folding pathway disappear. Simulations mimicking the folding of protein without disulfide bonds under oxidative conditions show that the native disulfide bonds are formed as the protein folds, indicating that folding guides the formation of disulfide bonds. The sequence of formation of the disulfide bonds is Cys64–Cys80 → Cys76–Cys94 → Cys30–Cys115 → Cys6–Cys127. Any disulfide bond that forms before its precursor in the sequence has to break and follow the sequence for the protein to fold. These results show that lysozyme also serves as a very good model system to probe the role of disulfide bonds and topological frustration in protein folding. The predictions from the simulations can be verified by single-molecule fluorescence resonance energy transfer or single-molecule pulling experiments, which can probe heterogeneity in the folding pathways.