Double Domain Swapping in Human γC and γD Crystallin Drives Early Stages of Aggregation
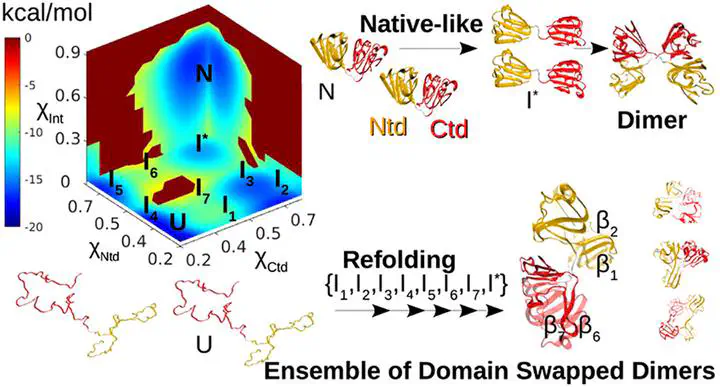 Image credit: B. Mondal
Image credit: B. Mondal
Abstract
Human γD (HγD) and γC (HγC) are two-domain crystallin (Crys) proteins expressed in the nucleus of the eye lens. Structural perturbations in the protein often trigger aggregation, which eventually leads to cataract. To decipher the underlying molecular mechanism, it is important to characterize the partially unfolded conformations, which are aggregation-prone. Using a coarse grained protein model and molecular dynamics simulations, we studied the role of on-pathway folding intermediates in the early stages of aggregation. The multidimensional free energy surface revealed at least three different folding pathways with the population of partially structured intermediates. The two dominant pathways confirm sequential folding of the N-terminal [Ntd] and the C-terminal domains [Ctd], while the third, least favored, pathway involves intermediates where both the domains are partially folded. A native-like intermediate (I*), featuring the folded domains and disrupted interdomain contacts, gets populated in all three pathways. I* forms domain swapped dimers by swapping the entire Ntds and Ctds with other monomers. Population of such oligomers can explain the increased resistance to unfolding resulting in hysteresis observed in the folding experiments of HγD Crys. An ensemble of double domain swapped dimers are also formed during refolding, where intermediates consisting of partially folded Ntds and Ctds swap secondary structures with other monomers. The double domain swapping model presented in our study provides structural insights into the early events of aggregation in Crys proteins and identifies the key secondary structural swapping elements, where introducing mutations will aid in regulating the overall aggregation propensity.